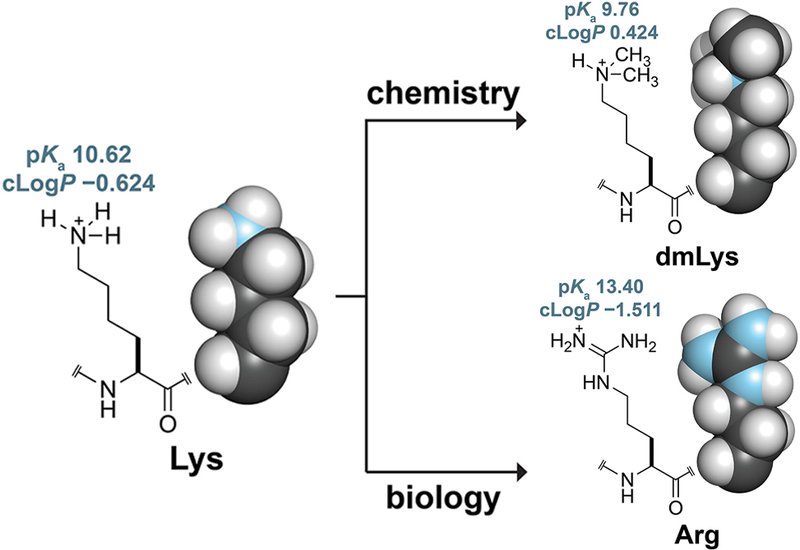
Lysine residues sit at the center of protein engineering strategy. Researchers modify them to block ubiquitination, improve crystallization, and dissect catalytic mechanisms. The canonical solution is site-directed mutagenesis to arginine, a substitution that preserves positive charge but introduces a planar guanidinium group with a pKa roughly 2.8 units higher than the lysine ammonium, five hydrogen-bond donors instead of three, and a larger van der Waals surface that can perturb protein–protein interfaces. The approach also requires separate mutagenesis rounds for each target residue and, critically, cannot address the N-terminal α-amino group, itself a substrate for ubiquitination. A chemical method that modifies every primary amine simultaneously, adds minimal steric bulk, and leaves overall charge intact would fill a genuine gap in the protein engineer's toolkit.
Researchers in the Raines Group at Massachusetts Institute of Technology, published in the Journal of Peptide Science, characterized reductive methylation as that chemical alternative. The reaction treats a protein with formaldehyde and a mild borane·dimethylamine reductant at 4°C in aqueous phosphate buffer at pH 7.5, converting each primary amine to a dimethylamino group through sequential imine formation and reduction. Only six atoms are added per site. The team used human ribonuclease 1, RNase 1, and QBI-139, a cytotoxic variant engineered to evade the endogenous ribonuclease inhibitor, RI, as model proteins. Together, these two proteins present nine modifiable amino groups each, including three lysine residues integral to RNA catalysis and a well-characterized protein–protein interaction surface.
Mass spectrometry confirmed complete labeling of both proteins, with the expected +252 Da shift corresponding to N,N-dimethylation of all nine sites. Thermostability, measured by differential scanning fluorimetry, shifted by no more than 1.5°C for either protein. The protein–protein interaction between dmQBI-139 and RI was preserved, with the dissociation constant rising from 0.64 ± 0.08 nM for unmodified QBI-139 to 1.8 ± 0.3 nM for the dimethylated form, a roughly threefold change that the authors attribute to flexibility of interfacial lysine residues rather than structural disruption. Bioconjugation chemistry was unaffected: an engineered cysteine at position 19 remained fully reactive after global dimethylation, enabling sequential thiol–maleimide coupling and strain-promoted azide–alkyne cycloaddition to append a HiBiT split-luciferase fragment for cellular tracking.
Catalytic activity told a different story. The kcat/KM for RNase 1 fell from 3.10 × 106 to 1.62 × 104 M−1s−1 upon dimethylation, a retention of only 0.14% of wild-type activity, consistent with the known catalytic roles of Lys7, Lys41, and Lys66. For QBI-139, the reduction was more pronounced, with kcat/KM dropping from 1.73 × 105 to 6.0 × 101 M−1s−1. This catalytic loss translated directly to cytotoxicity: QBI-139 showed an IC50 of 2.3 ± 0.2 μM against K-562 leukemia cells, while dmQBI-139 showed no measurable cytotoxicity in the same cell line. In HAP1 cells, a near-haploid leukemia line, dmQBI-139 did retain detectable toxicity with an IC50 of 301 ± 43 μM compared with 6.5 ± 1.3 μM for the unmodified protein. In HEK 293 AAV LgBiT cells, both the dimethylated and unmodified HiBiT conjugates entered cells at comparable levels after 24 hours and persisted with negligible signal decay over 72 hours, in contrast to a free HiBiT peptide control that showed measurable loss over the same period.
The pattern that emerges, preservation of global stability and binding properties alongside substantial disruption of active-site catalysis, positions reductive methylation as a practical strategy for applications that depend on protein stability and target recruitment rather than enzymatic turnover. The authors highlight biological proteolysis-targeting chimeras, bioPROTACs, as a direct use case, where intracellular degrader efficacy depends on binding affinity rather than catalysis, and where blocking ubiquitination of the degrader itself could prolong intracellular half-life. The demonstrated compatibility with orthogonal bioconjugation workflows further supports integration of reductive methylation into multistep protein engineering pipelines. The study also identifies a route to site-specific re-introduction of a single amino group on a fully protected scaffold: global dimethylation followed by elaboration of an engineered cysteine to γ-thialysine, a lysine isostere, via S-alkylation with 2-bromoethylamine.